
Pfizer and Moderna Analysis Re-do
Peter Doshi and colleagues' heroic attempt at re-analysis of the Phase 3 trials


There has been a bit of a stir lately concerning the posting at the beginning of this month of a pre-print (not yet peer reviewed) article entitled “Serious Adverse Events of Special Interest Following mRNA Vaccination in Randomized Trials”.
Words like “bombshell study” have been used to describe the findings. That sounds pretty significant, and certainly got my attention. With BMJ Senior Editor Dr. Peter Doshi as the senior author, could be! Dr. Doshi has a well earned reputation for telling inconvenient truths. So, let’s take a look.
Here are the headline results from the abstract:
Pfizer and Moderna mRNA COVID-19 vaccines were associated with an increased risk of serious adverse events of special interest, with an absolute risk increase of 10.1 and 15.1 per 10,000 vaccinated over placebo baselines of 17.6 and 42.2 (95% CI -0.4 to 20.6 and -3.6 to 33.8), respectively.
Combined, the mRNA vaccines were associated with an absolute risk increase of serious adverse events of special interest of 12.5 per 10,000 (95% CI 2.1 to 22.9).
The excess risk of serious adverse events of special interest surpassed the risk reduction for COVID-19 hospitalization relative to the placebo group in both Pfizer and Moderna trials (2.3 and 6.4 per 10,000 participants, respectively).
And the abstract discussion section:
“The excess risk of serious adverse events found in our study points to the need for formal harm-benefit analyses, particularly those that are stratified according to risk of serious COVID-19 outcomes such as hospitalization or death.”
The headlines look pretty serious at first glance. But the discussion section should alert us that the authors are being cautious. The authors are not signaling “pants on fire” findings.
What is really going on here? To understand this, a good place to start is this wonderfully clear and accurate summary of the initial Pfizer trial results from the Canadian COVID Care Alliance (posting which is apparently the sin that got me kicked off of Twitter and Linked-In last December, resulting in my becoming disconnected from about 600,000 followers).
You can also find a PDF summary of this analysis and findings here.
The bottom line is that the Pfizer Phase 3 trial which was used by NIAID, FDA and CDC to justify the emergency use authorization is pretty much a junk clinical trial which was inappropriately halted long before it even got close to meeting the intended follow up period, did not provide a sufficiently long follow up analysis of vaccination-associated adverse events, and in which the control group was intentionally eliminated. This resulted in basically erasing any opportunity to ever get to the bottom of what the major true risks of the Pfizer mRNA inoculations were. In terms of more minor risks, the study was not powered (not big enough) to evaluate those.
Into the breach, an intrepid group of (mostly) senior academic researchers have stepped forward. The expression “fools rush in where angels fear to tread” comes to mind, in that it has become extremely risky for any academic to question the approved vaccine narrative. But bravely this decidedly un-foolish group has stepped forward.
To my reading, the approach that they have taken with this analysis and report has been to make a good faith effort to perform the analysis of the Phase 3 clinical trials (those are supposed to be the “big, final” clinical trials prior to licensure of a product) which should have been performed by Moderna and Pfizer. Basically, the analysis that the FDA should have done themselves, and also should have forced Moderna and Pfizer to do. If White House Chief of Staff Mark Meadows had not put pressure on the FDA, perhaps it would have done the right thing. But it apparently caved and did not do it’s job, and here we are.
Herein lies the rub. The FDA not only did not do its job, but neither FDA nor Moderna nor Pfizer will release the primary data, which means that no-one else can do it either. As the authors of this recent analysis note in their discussion:
A systematic review and meta-analysis using individual participant data should be undertaken to address questions of harm-benefit in various demographic subgroups. Full transparency of the COVID-19 vaccine clinical trial data is needed to properly evaluate these questions. Unfortunately, well over a year after widespread use of COVID-19 vaccines, participant level data remain inaccessible.
Doshi and colleagues have repeatedly called for full disclosure in two prior publications, to no avail. So unless the data are included in the court mandated data release, the analysis which they perform in the current pre-print report may be as good as we are going to get. For further on this, please see
Tanveer S, Rowhani-Farid A, Hong K, Jefferson T, Doshi P. Transparency of COVID-19 vaccine trials: decisions without data. BMJ Evid Based Med [Internet]. 2021 Aug 9
Doshi P, Godlee F, Abbasi K. Covid-19 vaccines and treatments: we must have raw data, now. BMJ [Internet]. 2022 Jan 19;376:o102.
As Dr. Doshi and colleagues appropriately note,
“In 2013, the US and European industry trade organisations endorsed a joint statement on clinical trial data sharing, making a series of commitments that ‘recognise the importance of sharing clinical trial data in the interest of patients, healthcare and the economy”6 In 2015, the US Institute of Medicine similarly endorsed benefits of sharing clinical trial data, emphasising that ‘verification and replication of investigators’ claims’ were essential to the scientific process, and noting the numerous benefits to stakeholders ‘including payers of healthcare as well as patients, their physicians and researchers.’”
But if wishes were horses, beggars would ride. Pfizer and Moderna and the FDA clearly have no intention of heeding the pleas of the Senior Editor of the British Medical Journal, unless forced to do so by the US courts, and even then they will drag their heels for as long as possible. I can’t imagine why <sarcasm>.
The approach that Dr. Doshi and colleagues have taken is to rigorously cobble together a data set which is as close as possible to what might be the original by combing through the individual companies (“sponsors”) academic publications, as well as FDA and Health Canada websites for whatever tables or listings of adverse events could be gleaned from public presentations, and then assembled them to form the closest approximation to the “real” primary data that they could, and then analyzed those data sets.
In addition to journal publications, we searched the websites of the FDA (for advisory committee meeting materials) and Health Canada (for sections of the dossier submitted by sponsors to the regulator). For the FDA website, we considered presentations by both the FDA and the sponsors. Within each of these sources, we searched for SAE results tables that presented information by specific SAE type; we chose the most recent SAE table corresponding to the FDA’s requirement for a safety median follow-up time of at least 1 month after dose 2.
SAE is an abbreviation for serious adverse event. Note the last line - two months after the second dose. We know from the Cell paper last January that both the synthetic mRNA that is not really mRNA lasts for at least 60 days, as does the spike protein produced from that mRNA, so the “drug” is still present for at least two months after dose 2. Probably would be much better if the FDA insisted that the follow up for SAE were longer than one month. But they were in a rush because Trump’s Chief of Staff was telling them to get it done. So there it is. Cause and effect.
Getting back to the paper, to perform their analyses on the adverse event data which they were able to glean, Doshi and colleagues applied a list of “adverse events of special interest” (AESI) which had been compiled by CEPI and the Brighton Collaboration, and then approved by WHO. This list had been compiled before the trial started. Now in retrospect, we have Pfizer’s extensive table of potential AESI which appears to have been compiled AFTER the emergency use authorization was enacted, and the authors could have used that. But Doshi et al are being real Boy Scouts, and chose to only evaluate the AESI list which existed prior to the trial data becoming available for their analyses, in their apparent attempt to retrospectively do what should have been done originally.
The problem with this is that they do not actually have access to the patient level data, so they have had to make some assumptions about those primary data, particularly in terms of their numeric/statistical distribution.
“Another limitation is our lack of access to individual participant data, which forced us to use a conservative adjustment to the standard errors. The 95% CI calculated are therefore only approximate because we do not know which patients had multiple events. Furthermore, despite our attempt to remove efficacy endpoints from our analysis (i.e., SAEs labeled as COVID-19, COVID-19 pneumonia, and “SARS-CoV-2 test positive”), it was not possible to identify and remove SAEs that occurred in patients with serious complications of COVID-19 (e.g., acute respiratory failure, cardiac arrest, and acute kidney injury), which are common.”
In other words, they did the best that they could, but had to include some assumptions.
Here is the key data table which resulted from all of this hard work:
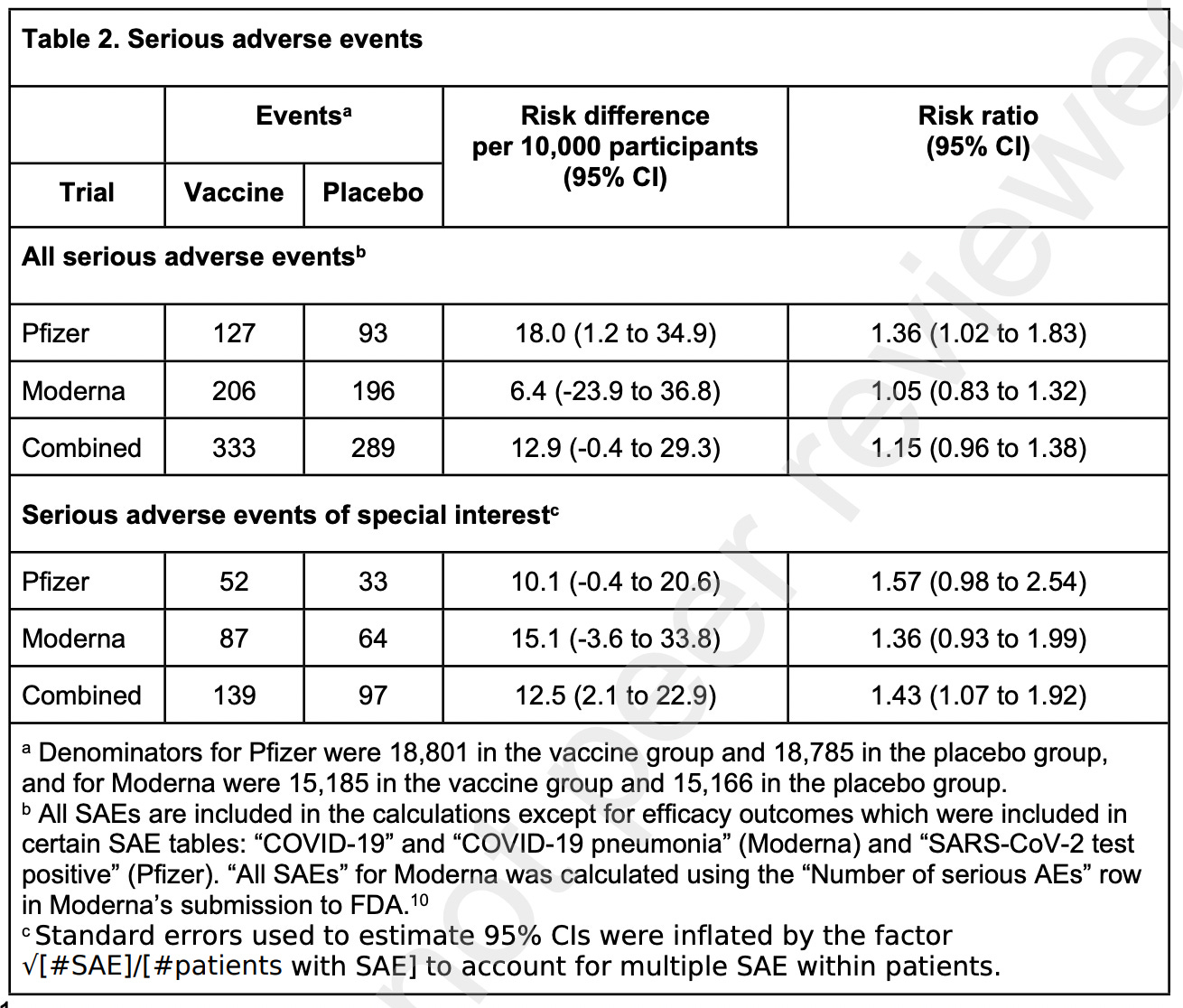
Notice the risk ratio columns, and in particular, the 95% confidence interval (abbreviated as CI). A risk ratio where the control group and the experimental group are equivalent would be 1.0. Greater than 1.0 (in this case) would mean that there was more risk of adverse events to those receiving vaccine. But there is a statistical range around that number (when randomly testing, and setting the statistical test threshold that 95 out of 100 times the result will fall within that range). So, if the confidence interval spans less than one to greater than one, you cannot conclude that there is a statistical difference between results for control and vaccine treated. As is the case with many of these tests. Now they are all pretty skewed to very close to 1 and greater than 1. So that suggests that if the number of patients tested had been larger, they might well all reach statistical significance. But this is actually a modest sample size for a Phase 3 vaccine trial. Again, the FDA let the sponsors get away with this, but these are the data which are available. And there is no way we can ever get back to that point in time, because almost everyone has either been vaccinated or infected at the present.
In vaccine research, for purposes of estimating study sample size, we apply the rule of three. If you want to reliably detect an adverse event that occurs at the rate of once every thousand patients, you should test 3,000 patients in your vaccinated group. So in the case of Pfizer trial, it is powered to detect adverse events that occur about once in (18,800/3) = 6,266 patients. Moderna, (15,185/3) = 5,061 patients. Adverse events occurring less frequently than that would generally not be detected at a statistically significant level. Correcting for frequency of adverse events randomly occurring in the control group, and normalizing to event # per 10,000 patients gets you to the data summarized in the table.
Also note that, for the Serious Adverse Events of Special Interest, to get to what their estimates indicate would be statistical significance, they had to combine the data from the Pfizer and the Moderna clinical trials. Something that would never be done in a “real world” setting, as the two products are different, involve different delivery formulations, and are administered at very different doses of mRNA.
From the above, you can appreciate that this analysis is pretty much as good as can be had, given the what the authors have had to work with. But now you can also appreciate why they (appropriately) concisely reported their findings (“All we want are the facts, ma’am”), and then drew an appropriately cautious conclusion:
“The excess risk of serious adverse events found in our study points to the need for formal harm-benefit analyses, particularly those that are stratified according to risk of serious COVID-19 outcomes such as hospitalization or death.”
This was a heroic collaborative effort to try to get back to a point in the clinical research history of these mRNA vaccines when critical decisions were made which would quite literally impact on the course of history. The decisions at that time were rushed, the usefulness of the two studies destroyed (intentionally?) by stopping the studies prematurely and then vaccinating the control group, and what data were gathered have been largely hidden from those who wish to do independent analyses. The authors of the current analysis re-do attempt have done their best. But, as Dr. Doshi and colleagues have repeatedly requested, the proper analysis cannot be performed unless the original data sets are released.
Until then, we’ll always have Paris. Here’s looking at you, kid.
Play it again, Sam. Play “As time goes by”.

We need a serious lawyer and a serious lawsuit and some serious expert witnesses. Enough is enough
Combine this with Robert Kennedy's Children's Health Defense FOIA showing the CDC didn't even analyze safety signals from the VAERS data, it looks grim. The CDC probably knows just as well as Pfizer and the FDA that the data has been warped not to damage the bottom line (best case scenario) or this vaxx is being used as a massive experiment with EUA to develop more moneymakers, or it's a bioweapon (worst case scenario). In any case they are hiding the data to cover their little cushy tushies because if it was incompetence, their would be data going both ways making the experimental mRNA injection look good or bad, but the data is warped to make it all look good.
It's all done with our tax money too. Joy. Can we defund all these horrid 3 letter agencies, please?
https://childrenshealthdefense.org/defender/cdc-vaers-covid-vaccine-safety/