
Prasad FDA Memo: Regulatory Implications
Yes, the investigation of VAERS childhood deaths is important. But Media is overlooking the big picture.


I spent my early Monday hours today doomscrolling through “X”, monitoring the outrage and consternation concerning FDA/CBER director Dr. Vinay Prasad’s “official” (actually unofficial “leaked”) acknowledgment that there have been COVID mRNA-induced vaccine deaths of children. Yes, I guess that is an important event, but I first became aware of these specific findings last summer in the context of my service at ACIP. So, it's pretty much yesterday’s news for me.
Because of how I first encountered the data and investigation findings, I am embargoed by FACA federal law from disclosing what I know of them. However, I can assure you that the findings of Dr. Tracy Beth Hoeg (FDA Office of the Commissioner, ACIP FDA liaison) are much more shocking than just the number and age of the vaccine-related deaths.
In reviewing the social media posts and chatter, what seems to be almost completely overlooked are the Regulatory policy components of this “leaked” internal memo from Director Prasad to his bureaucracy. When I wrote my initial essay last Saturday posting the letter with a brief introduction (titled “A Revolution in Vaccine Regulation and Approval”), it was the public policy changes being announced that struck me as revolutionary. I have been alerting parents to the risks of harm to children from the genetic COVID vaccines for many years now. Not surprisingly, I experienced a wave of concerted attacks for this from corporate media, internet trolls/bots, and even the Israeli and Spanish governments! But I stand by every word, and have been vindicated and validated since (including by this recent memo from CBER Director Dr. Prasad). The fact that pediatric deaths have occurred post coerced and mandated inoculation with these products is not news to me.
Last year, when quietly serving as an expert witness in a large “mandates” case filed against the largest school district in the USA, I made the point that the district had mandated children and employee use of a product and medical procedure that had death as a side effect. At that point, the defense basically folded and settled the case out of court. There is no way to mitigate the impact of that truth bomb on a jury. There is no credible defense against the fact that children (and adults) were required to receive an injected product that had death as a known side effect - not to mention myocarditis. Both of which side effects we now know were covered up by US Federal Officials. As Steve Kirsh has pointed out on “X”, this cover up appears to be a prosecutable offense. Lets park that issue for now, delegating over to AG Pam Bondi.
Dr. Maryanne Demasi has done a great job of summarizing the publicly available information and timeline on Dr. Hoeg’s analysis and subsequent internal FDA backlash . Unfortunately, if you wish to read her Substack journalistic analysis, it is behind a paywall, so you will need to subscribe.
Moving beyond the outrage, PsyWar/propaganda backlash from the captured media and industry (and academic) vaccine shills, and general gnashing of teeth over the obvious fact that children were damaged and killed by these products, I think the larger, unreported story is the Regulatory policy changes outlined in the Prasad memo.
Since this has not been well covered by others, I have chosen that as my topic for today. Like so much of what I write and publish these days, this has a dual purpose: both to inform our readers and to help me personally sort out observations and implications. What Dr. Prasad has outlined will have a profound impact on ACIP positions and recommendations to the CDC Director. I suspect it will also transform (revolutionize) the entire vaccine industry, unless it gets blocked from implementation in some way.
As ACIP vice chairperson, I will need to help facilitate analysis and advice on this topic to the Director. Seeking to be as objective as possible given the topic, let’s get into the weeds on this.
Regulatory Implications
Starting with Paragraph #2 of the letter:
Prior to joining the US FDA, the FDA Commissioner closely followed reports of vaccine-induced myocarditis. Unlike the COVID virus, which has a steep age gradient-- being at least 1000 times more likely to kill an 80 year old than an 8 year old-- myocarditis appeared to have the opposite pattern. Young, healthy boys and men-- those least likely to experience bad covid outcomes-- bore the greatest risk. The risk was as high as ~200-330 per million doses given in the highest risk demographic groups. Notably, the US FDA and CDC were not the first to recognize the safety signal-- instead the Israelis were-- and worse in May of 2021, then CDC director Rochelle Walensky stated, “We have not seen a signal and we’ve actually looked intentionally for the signal in the over 200 million doses we’ve given,” Many felt this statement was dishonest and manipulative.
Prasad is not referencing a specific regulated product, but rather seems to refer to the general class of COVID vaccine products. Personally, I object to and disagree with this approach. Each product is unique, and should be addressed as such. General statements about a class of products (“COVID vaccines”) are not appropriate from a regulatory science perspective. I advocate adopting this position at both FDA and CDC.
Prasad is describing an official CBER position that the risk of myocarditis post administration of these product is at least 200-330/million doses (2-3.3/ ten thousand) in this highest risk cohort. Nuance here being doses. That means for a patient receiving the initial vaccine series (two doses), there is a risk of 4-6.6 myocarditis events/ten thousand vaccinees (0.04 to 0.066%). For comparison, current official COVID case rates of death in children (CFR) are in the range of 0.04–0.2%. Also keep in mind that myocarditis is one of many adverse events known to be associated with receipt of these products.
Prasad points out that former CDC director Walensky lied and thereby suppressed public disclosure of what was then known to be a myocarditis serious adverse event.
Putting these facts together, it is horrifying to consider that the US vaccine regulation, including our actions, may have harmed more children than we saved.
It is now the official position of the FDA/CBER that USG vaccine policies may have caused more harm than would have been associated with allowing SARS-CoV-2 to infect children without vaccination. This overlooks the additional impact of active FDA and USG suppression of the early treatment options pioneered by front-line physicians all over the world.
As a professor, I agreed with Gruber and Krause. Furthermore, there have been prior CBER directors who have held this chair and had fundamentally different views. Some have felt the CBER director should override reviewers to approve gene therapies that do not work because of patient demand. When these products later result in post market deaths, it is difficult to take corrective action. I favor approving products with benefits that exceed risks.
This is a rather pointed criticism of the preceding FDA/CBER Director Dr. Peter Marks. Marks, together with HHS ASPR Robert Kadlec, initially formulated the “Operation Warp Speed” plan.
Prasad appears to be implicitly acknowledging that the mRNA and adenoviral vectored COVID vaccine products are, in fact, gene therapies. If so, this is a profound repudiation of the Marks’ position, and would imply that the gene therapy guidance to industry would apply to these vaccine products. That would be a major shift, requiring issues such as shedding to be addressed prior to authorization of human clinical trials. He is also broadening the topic out to gene therapies in general.
Prasad appears to be taking the position that a more rigorous, traditional risk/benefit -based approach to regulatory authorization be employed for all of these products. If I am interpreting this correctly, this policy shift will profoundly impact on cost and product development timelines for all gene therapy products including all gene therapy technology-based vaccines.
In the immediate future, this shift is likely to significantly impact on self-replicating RNA vaccine regulatory pathways as well as on any new modified (or otherwise) mRNA-based vaccine submissions.
Covid-19 vaccines earned 100 billion dollars globally. The annual US vaccine market is estimated to be over 30 billion dollars, projected to pass 50 billion in a decade, and a single new vaccine for pregnant women has industry analysts estimating 1 billion a year in annual returns. Additionally, vaccines do not go “generic.” There is no biosimilar pathway. You can’t show your biosimilar vaccine has the same antibody titer and get approval. This means two things: companies can expect long tails of earnings, and FDA acknowledges that cell and humoral immunity surrogates are insufficient for generic approvals-- a position I agree with. The fact that we don’t offer generic or biosimilar vaccines because no amount of cell or humoral mediated immune surrogates would mean that a product retains efficacy has a deeper logical conclusion: how can we accept such endpoints to approve entirely novel products?
Prasad is acknowledging that the unique nature of vaccine products and the associated regulatory positions (including CDC VFC policies, by the way) results in a highly profitable virtual long-term monopoly for these products.
The impact of second clause in this paragraph is revolutionary. Cell and humoral immunity surrogates are insufficient for vaccine approvals. Surrogates are not the same as validated correlates of protection, as exists for influenza vaccines. Validated correlates of protection do not exist for COVID vaccines. The implications are multifold.
First, humoral immunity surrogates (notoriously mouse based) were used to authorize the COVID “booster” products.
This implies that immune surrogates will no longer be accepted for these products. One interpretation could be that the prior product authorization is no longer valid, and the related COVID vaccine products should no longer be marketed.
If surrogates (unproven correlates of protection) will not be accepted for regulatory authorization for vaccines, the implication is that actual well-controlled clinical trial data demonstrating efficacy/effectiveness will be required for licensure/marketing authorization for vaccines.
Our general approach in CBER will be to direct vaccine regulation towards evidence based medicine. This means: we will take swift action regarding this new safety concern, we will not be granting marketing authorization to vaccines in pregnant women based on unproven surrogate endpoints (any prior promises will be null and void), and we will demand pre-market randomized trials assessing clinical endpoints for most new products. Pneumonia vaccine makers will have to show their products reduce pneumonia (at least in the post-market setting), and not merely generate antibody titers. Immunogenicity will no longer be used to expand indicated populations-- these populations should be included in premarket RCTs.
“We will take swift action regarding this new safety concern” refers to the pediatric death adverse event now associated with the COVID vaccine products.
At a minimum, this implies a “black box” warning of death, in addition to the current myocarditis warning. It suggests a high likelihood of rescinding FDA authorization for use of these products in children and young adults.
“we will not be granting marketing authorization to vaccines in pregnant women based on unproven surrogate endpoints (any prior promises will be null and void)” implies that current or pending FDA authorization for use of biologic products (including vaccines) will be rescinded unless they meet the new criteria of clear demonstration of safety and efficacy/effectiveness in pregnancy. This is basically a return to standard regulatory policy prior to Dr. Peter Marks’ tenure.
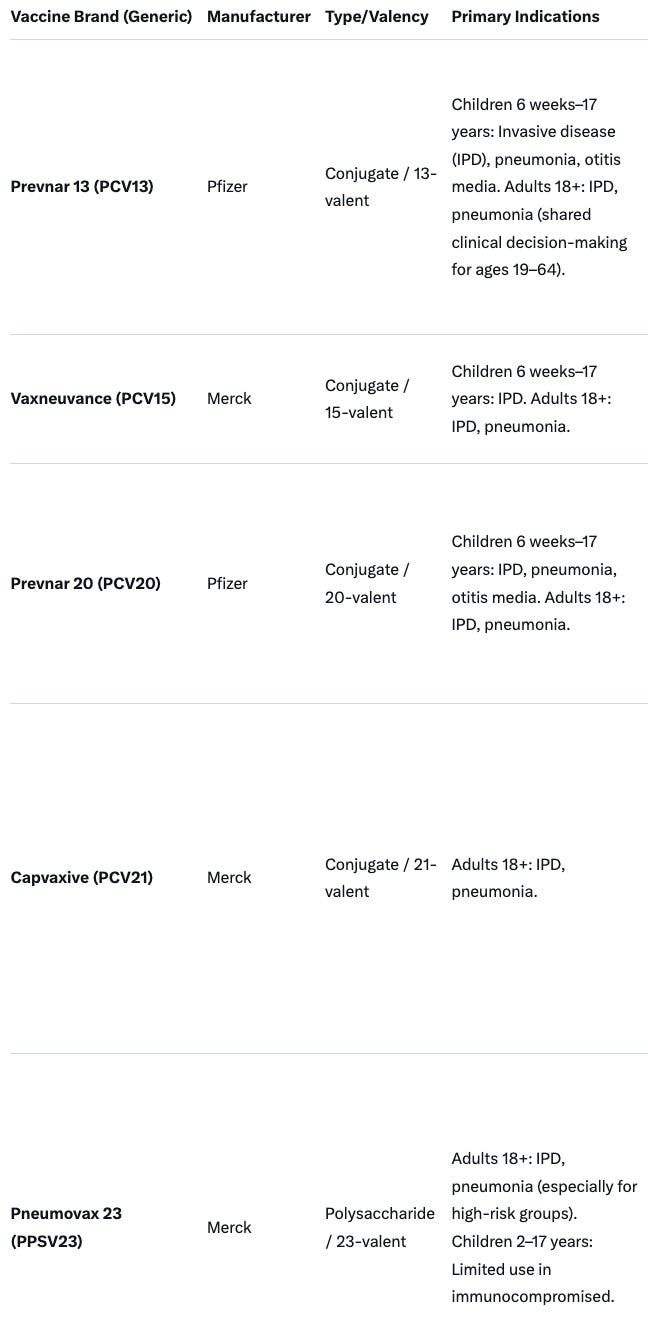
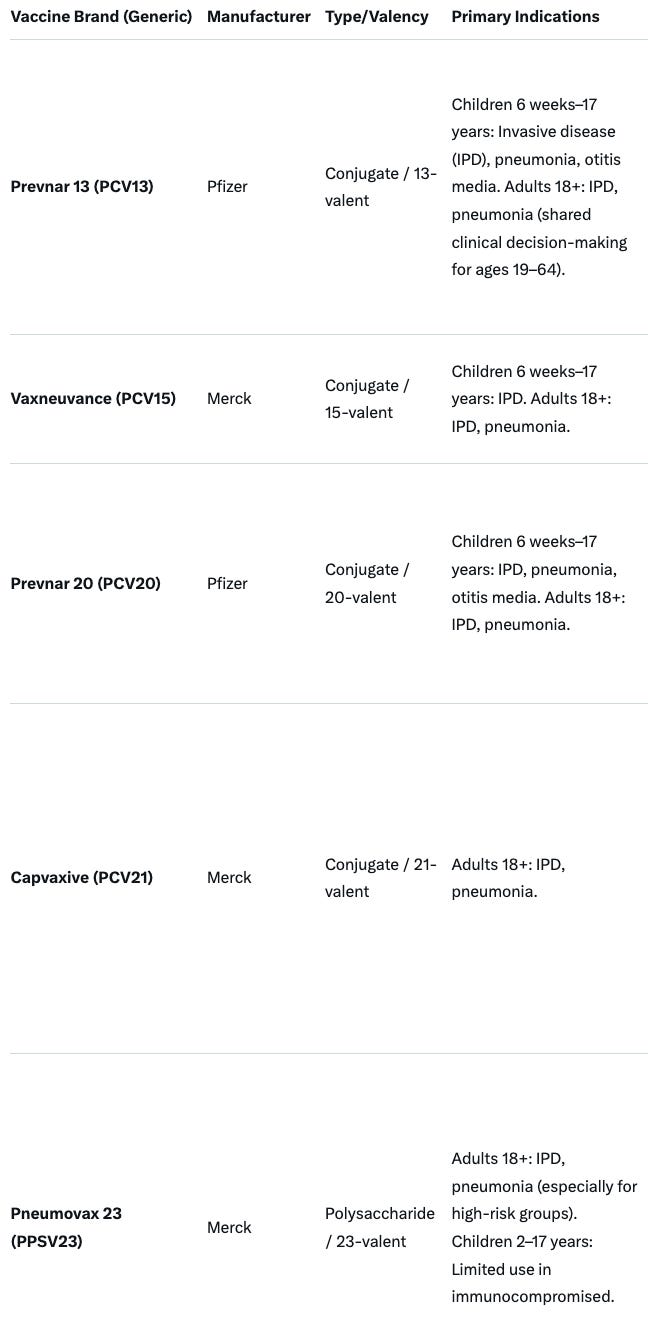
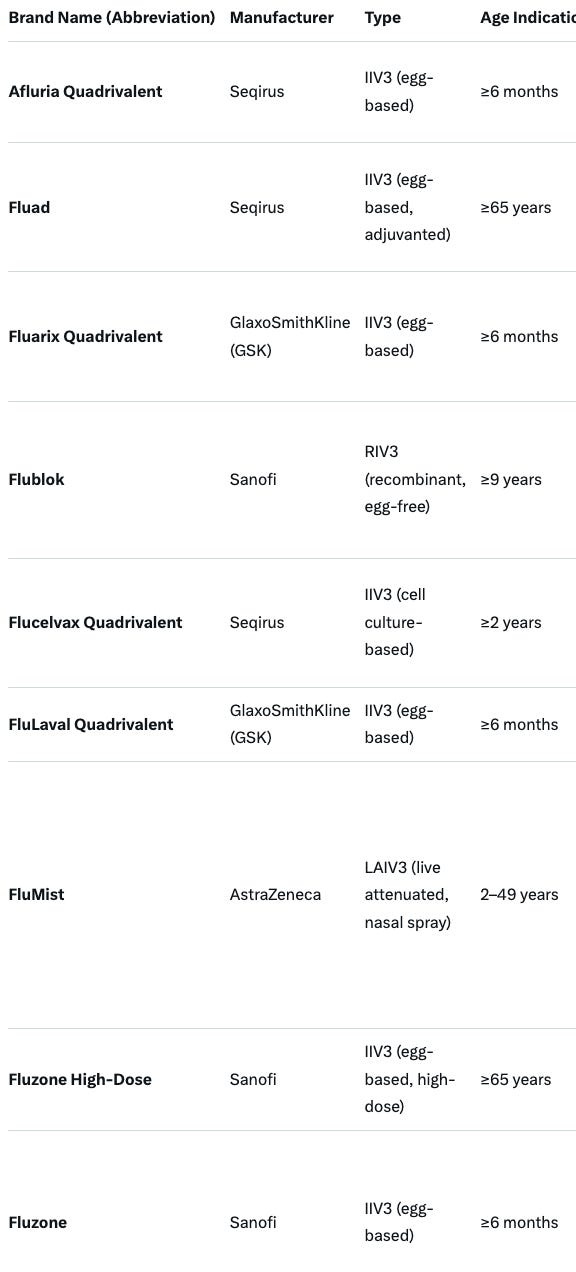
“Pneumonia vaccine makers will have to show their products reduce pneumonia (at least in the post-market setting), and not merely generate antibody titers. “ Businesswise, this will be a threat to both Pfizer and Merck vaccine estates. Relevant products that must meet these new requirements are summarized below. ACIP will need to reevaluate current recommendations for each of these products:
.
We will revise the annual flu vaccine framework, which is an evidence-based catastrophe of low quality evidence, poor surrogate assays, and uncertain vaccine effectiveness measured in case-control studies with poor methods. We will re-appraise safety and be honest in vaccine labels.
I cannot overstate the importance and impact of this brief statement. In the context of vaccination regulatory policy, this is truly revolutionary. Sacred cows previously not allowed to be questioned or examined are suddenly being lined up for slaughter. I have previously been fired from vaccine companies or clients for questioning many, if not all, of these issues.
The CBER director is questioning the one “validated correlate or protection” that exists in the vaccine industry - the antiquated and poorly predictive hemagluttination (HA) titer assay.
Prasad is criticizing “uncertain vaccine effectiveness measured in case-control studies with poor methods”. This is a direct criticism of the work of the CDC influenza monitoring system. FDA requires efficacy studies. CDC performs effectiveness studies. The CBER director is stating that the CDC influenza monitoring network employs “poor methods”.
“We will re-appraise safety and be honest in vaccine labels.” This is code for saying that annual (and pandemic) influenza vaccine labeling (specifically package inserts) have been misleading and have historically not provided an accurate assessment and disclosure of safety risks.
The annual influenza vaccine market in USA generates annual sales of 4.5-5 billion USD for about 148 million doses (average about 34 dollars per dose).
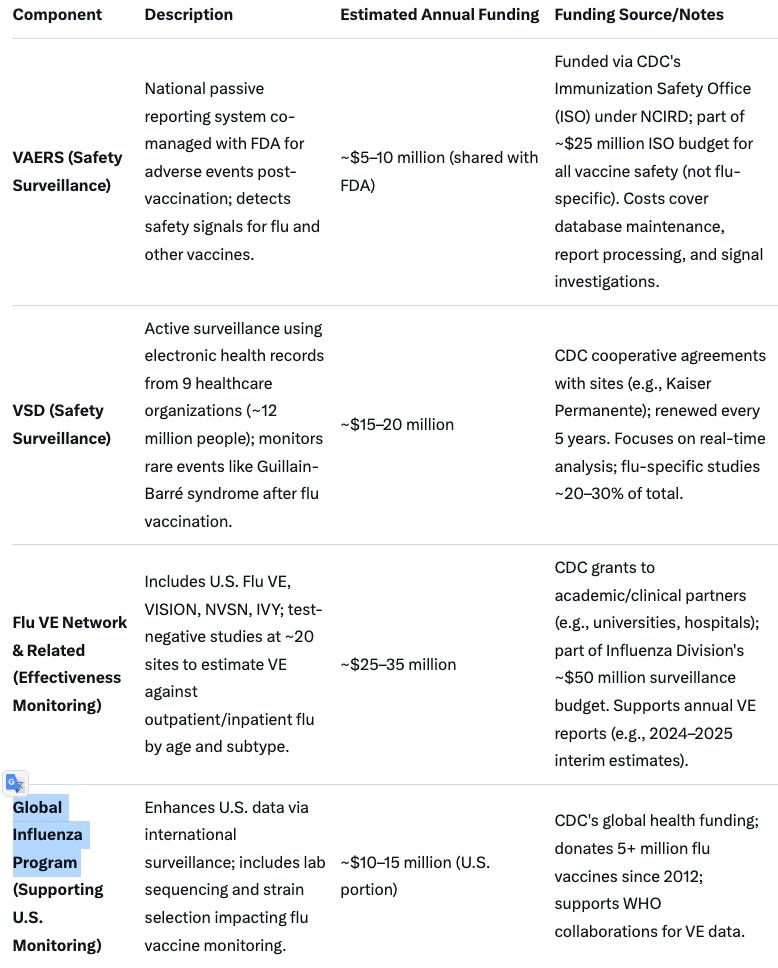
These policy changes will impact on a wide range of products and manufacturers. This will also require ACIP review and consideration of each product and the regulatory implications, as well as reevaluation of the CDC influenza vaccine monitoring program. This involves the U.S. Flu VE, VISION, NVSN, IVY; test-negative studies at ~20 sites to estimate VE against outpatient/inpatient flu by age and subtype (25 - 35M$ per annum), the VSD system (15-20M$) as well as the Global Influenza Program (10-15M$ annual for US portion of expense).
Also relevant is the impact of these policy shifts on influenza vaccine mandates, including in schools, hospitals, Universities, Medical Schools, and the US Military. The ripple effect will be widespread.
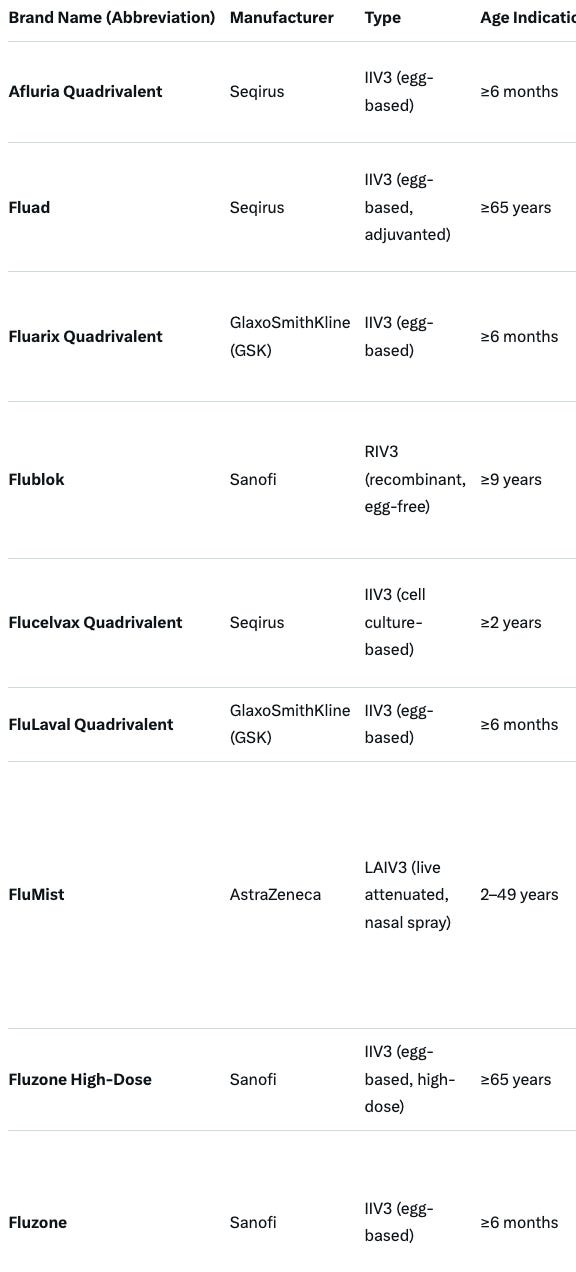

In Conclusion
This is the most comprehensive shift in vaccine industry regulatory policies in my lifetime and over 30 years of experience working in this industry. As FDA and CBER have traditionally set the standard for global vaccine regulatory policies, this is likely to have ripple effects across the European Union (European Medicines Agency), Canada, Great Britain, Australia, and New Zealand.
In terms of the vaccine industry, this memo indicates profound changes. In order for these to be sustainable, in terms of policy, these comments from the Director will need to be translated into changes in the official “Guidance for Industry” documents that serve as guidance for all relevant FDA regulatory actions and communications. This may take two or more years, because of the required review and comment periods. However, these policy positions are likely to become “soft” mandates until then, so long as Dr. Prasad remains CBER director. You can expect that Pharma, captured academics, and their media surrogates will mount an aggressive propaganda campaign aimed at discrediting Dr. Prasad and these policies. In many ways, this has already begun with the propaganda and spin around the assertion that the VAERS mortality report analyses are invalid.
Beyond this, the geopolitical implications are also profound. The UK has determined that COVID vaccine-associated death data are not to be disclosed. That position may not be sustainable now. We are in unchartered territory, and governments, the WHO, non-governmental organizations, and media will all be forced to account for their actions in promoting these products as well as their actions suppressing and ridiculing physicians and scientists that raised objections to the promoted false narratives.
"..governments, the WHO, non-governmental organizations, and media will all be forced to account for their actions in promoting these products as well as their actions suppressing and ridiculing physicians and scientists that raised objections to the promoted false narratives."
I certainly hope this proves to be true, but I have doubts.
FAFO continues! This is exactly what I voted for.