
AI Analysis Finds Natural Origins More Likely for Omicron Variant B.1.1.529
Highlights Potential of New Role for Artificial Intelligence in Biological Weapons Convention (BWC) Monitoring


AI Analysis Finds Natural Origins More Likely for Omicron Variant
Highlights New Role for Artificial Intelligence in Bioweapons Monitoring
A new analytical study applying artificial intelligence tools to the origins of the COVID-19 Omicron variant concludes that while a laboratory origin cannot be ruled out, the available evidence currently favors natural explanations.
The report applies a “six-layer” verification framework designed to evaluate potential violations of the Biological Weapons Convention (BWC), integrating genomic analysis, open-source intelligence, supply-chain monitoring, environmental epidemiology, behavioral indicators, and predictive modeling. Using Bayesian statistical methods and 10,000 Monte Carlo simulations, the analysis estimates a 24.6 percent probability that Omicron originated in a laboratory, compared with roughly 75 percent probability that it arose through natural processes, though researchers emphasize that the estimate remains highly uncertain.
This study continues the development and validation of an AI-powered six-layer analysis framework designed to monitor compliance with the International Biological Weapons Convention. The framework is being tested and improved using retrospective analysis of alleged laboratory incidents and bioweapons deployment claims to establish baseline accuracy before implementing real-time surveillance systems.
Previous case studies using this methodology have examined the SARS-CoV-2 Wuhan Institute of Virology laboratory-leak hypothesis, allegations that RSV entered human populations through an accident at a mid-Atlantic research facility, theories regarding the origins of Lyme disease in laboratories, claims about the Dugway Proving Ground’s role in live anthrax spore distribution incidents, and the theory that the currently circulating Avian Influenza virus (H5N1 Clade 2.3.4.4b) originated as a laboratory leak.
Omicron, first identified in southern Africa in November 2021, immediately drew scientific attention because of its unusually large number of mutations—roughly 50 relative to the original SARS-CoV-2 strain. Phylogenetic analysis also revealed an 18-month evolutionary gap between Omicron and its closest known viral relatives, raising questions about where the variant evolved during that period.
The study evaluates four major origin hypotheses discussed in the scientific literature. The most widely accepted theory is that the variant evolved during prolonged infection in an immunocompromised patient, where sustained immune pressure allowed the virus to accumulate many mutations over time. A second possibility involves transmission from humans into rodents followed by adaptation in an animal reservoir before re-entering the human population. Other explanations—including long-undetected spread in poorly monitored regions or laboratory experimentation—remain possible but lack definitive evidence.
Genomic analysis produced the strongest signal raising questions about Omicron’s evolutionary path. Researchers found unusually strong positive selection in the spike protein and a mutational pattern resembling adaptation to mouse cells. However, the analysis also found no genetic signatures of deliberate engineering, such as synthetic elements or codon-optimization artifacts.
Other layers of the investigation weakened the laboratory hypothesis. The report found no evidence of suspicious laboratory procurement patterns, unusual DNA synthesis orders, or identifiable institutional activity linked to the emergence of the variant. That absence of corroborating signals, the authors note, significantly lowers the likelihood that Omicron resulted from laboratory work.
Rather than offering definitive attribution, the study is designed as a demonstration of how artificial intelligence could assist international monitoring of biological weapons risks. By continuously scanning genomic databases, scientific publications, procurement records, and epidemiological signals, AI systems can identify anomalies that may warrant further investigation.
The approach reflects growing interest in Washington in using advanced data analytics to strengthen global oversight of biological threats. In recent remarks, both the President of the United States and State Department Undersecretary for Arms Control and International Security Thomas DiNanno have pointed to artificial intelligence as a potential tool for improving compliance monitoring under the Biological Weapons Convention, which prohibits the development and stockpiling of biological weapons.
DiNanno has argued that modern biotechnology increasingly leaves digital traces—from gene sequence databases to procurement records—that advanced analytical systems could detect and analyze at global scale. AI-driven monitoring platforms, he has suggested, could provide early warning signals of unusual biological activity long before traditional investigative mechanisms detect problems.
The Omicron analysis offers a retrospective example of how such systems might function. The framework flagged statistically unusual genomic features in the variant while simultaneously incorporating negative evidence from other domains, producing a cautious probabilistic estimate rather than a definitive conclusion.
Researchers emphasize that the results should not be interpreted as proof of any particular origin. Instead, they argue that the study illustrates how multi-layered AI analysis could help governments and international organizations prioritize investigations and strengthen transparency under the Biological Weapons Convention.
In the case of Omicron, the ultimate origin of the variant may never be conclusively determined. But the study suggests that the same analytical tools used to examine the past could play a critical role in identifying and monitoring biological risks in the future.
Sources
Raquel Viana et al., “Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa,” Nature (2022).
Kai Kupferschmidt, “Where Did ‘Weird’ Omicron Come From?” Science (2021).
Lok Bahadur Shrestha et al., “Evolution of the SARS-CoV-2 Omicron Variants BA.1 to BA.5,” Reviews in Medical Virology (2022).
Sandile Cele et al., “SARS-CoV-2 Prolonged Infection during Advanced HIV Disease Evolves Extensive Immune Escape,” Cell Host & Microbe (2022).
Haiwei Gu et al., “Evidence for a Mouse Origin of the SARS-CoV-2 Omicron Variant,” Journal of Genetics and Genomics (2022).
Mahmoud Kandeel et al., “Omicron Variant Genome Evolution and Phylogenetics,” Journal of Medical Virology (2022).
World Health Organization, “Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern,” WHO, November 26, 2021.
Office of the Director of National Intelligence, “Declassified Assessment on COVID-19 Origins,” U.S. Intelligence Community, 2023.
World Health Organization Scientific Advisory Group for the Origins of Novel Pathogens (SAGO), “Preliminary Report on Studies of the Origins of SARS-CoV-2,” WHO, 2022.
This article is based on a comprehensive analysis applying the AI-Enhanced BWC Verification Framework developed by Dr. Robert Malone to the hypothesis that the original SARS-CoV Omicron variant B.1.1.529 was developed in a laboratory environment rather than evolving naturally. The analysis was conducted using open-source intelligence, government documents, and scientific literature.
The opinions expressed herein are solely those of the author, and do not represent the opinions of the US Government, US State Department, the US Department of Health and Human Services, or the US Centers for Disease Control and Prevention.
SIX-LAYER BWC VERIFICATION ANALYSIS
Omicron B.1.1.529: Laboratory Engineering Hypothesis
Updated Assessment with Literature Review and Genomic Drift Analysis
PRIMARY RESULT
24.6% Laboratory Origin Probability
90% Confidence Interval: 4.6% to 53.6%
Monte Carlo N = 10,000 | Seed = 42 | Convergence sigma = 0.00018
Introduction: The Origin of Omicron B.1.1.529. A Review of the Scientific Literature and Competing Theories
Background and Discovery
On 24 November 2021, the Network for Genomics Surveillance in South Africa reported a novel SARS-CoV-2 variant to the World Health Organization. Designated B.1.1.529, the variant had been detected in samples collected from 8 to 16 November 2021 in both Botswana and South Africa, with the earliest confirmed specimens originating from Johannesburg on 8 November and from Botswana on 9 November. Two days later, on 26 November, the WHO classified it as the fifth Variant of Concern and named it Omicron, after the fifteenth letter of the Greek alphabet. By 6 January 2022, Omicron had been confirmed in 149 countries, and within weeks it had displaced Delta to become the globally dominant SARS-CoV-2 lineage, a transition unprecedented in its speed in the pandemic’s history.1,2
What immediately distinguished Omicron from its predecessors was not its transmissibility alone, but the sheer scale and novelty of its mutational profile. As of June 2022, Omicron carried approximately 50 mutations relative to the original Wuhan-Hu-1 reference genome, more than any prior SARS-CoV-2 variant. Thirty-two of these affected the spike protein, with 15 residing in the receptor-binding domain (RBD) alone.1 The combination of mutations conferred striking immune evasion properties: multiple studies documented an 8 to 127-fold reduction in vaccine efficacy against Omicron compared to the ancestral strain.4 It multiplied approximately 70 times faster than Delta in bronchial tissue but, paradoxically, appeared to cause less severe lower respiratory disease, a clinical dissociation that itself became a subject of intensive investigation.29
Critically, phylogenetic analysis revealed that Omicron did not evolve from any other circulating variant. Its closest relatives in global genomic databases dated to mid-2020. This represents a gap of some 18 months during which no intermediate sequences were detected across millions of GISAID submissions. This absence of an evolutionary trail, combined with the density and novelty of its mutations, immediately raised a question that has not been definitively resolved: where, and in what host environment, did Omicron evolve?3,7
The Central Scientific Problem
Omicron’s evolutionary history is unknown. Its mutational divergence from the next closest known sequences implies an extended period of evolution, estimated at 12 to 18 months, in a host environment not reflected in any sequenced viral population. Three principal natural hypotheses, and one contested laboratory hypothesis, have been advanced to account for this gap. None has been definitively confirmed.
Theory 1: Intra-host Evolution in an Immunocompromised Individual
Status: Most widely accepted among mainstream virologists. The leading hypothesis in the peer-reviewed literature is that Omicron evolved through prolonged SARS-CoV-2 replication in a single chronically infected, immunocompromised patient. This patient was most likely one with advanced HIV disease, haematological malignancy, or another condition causing profound B-cell or CD4+ T-cell dysfunction. In such patients, the immune system exerts sustained but sub-sterilising selection pressure on the virus: antibodies are generated but cannot clear infection, creating a prolonged evolutionary environment in which the virus accumulates immune-escape mutations over months to years.4,5
The hypothesis draws direct support from documented case series. Researchers have recorded chronic SARS-CoV-2 infections in immunocompromised individuals lasting from several months to over a year, during which the virus accumulated large numbers of spike mutations, including mutations later found in Omicron. A study of severely immunocompromised patients during the Omicron period found that four of five patients with very prolonged viral shedding accumulated consensus-level spike mutations, the majority in the receptor-binding domain.16 The HIV connection is geographically plausible: the initial Omicron sequences were obtained from an HIV-infected patient in Botswana, and South Africa and the surrounding region account for approximately half of the world’s population living with uncontrolled HIV infection, approximately 14% of South Africa’s total population.15 A 2025 phylogeographic study confirmed that Gauteng Province in South Africa likely played a central role in the emergence and amplification of multiple Omicron lineages, and directly linked uncontrolled HIV infections to the conditions that foster highly divergent SARS-CoV-2 evolution.14
The hypothesis is not without limitations. A Lancet Microbe prospective study of immunocompromised patients found that while spike mutations did accumulate in prolonged infections, the specific mutations acquired were mostly distinct from those defining Omicron, and none of the five patients with most prolonged shedding showed evidence of onward transmission based on placement in a global phylogenetic tree of over eight million sequences. The study concluded that while immunocompromised patients represent an important evolutionary reservoir, the path from chronic intra-host evolution to a globally spreading variant requires additional steps that are not fully characterised.16
Theory 2: Zoonotic Spillback from a Murine Host
Status: Scientifically supported by molecular evidence; not confirmed by surveillance. The second major natural hypothesis proposes that an early SARS-CoV-2 lineage jumped from humans to a rodent population, accumulated mutations adapted to the murine ACE2 receptor during sustained transmission in that animal reservoir, and subsequently spilled back into humans. The molecular evidence for this scenario is substantial and was first systematically documented in a preprint from the Chinese Academy of Sciences in December 2021, later published in the Journal of Genetics and Genomics.6
The core evidence is threefold. First, the mutational spectrum of Omicron’s 45 pre-outbreak point mutations was statistically different from the spectrum of viruses known to have evolved in human hosts (p = 0.004), but closely resembled the spectrum associated with virus evolution in a murine cellular environment. Second, the Omicron spike protein carried numerous mutations, including Q493K, Q498H, and N501Y, specifically associated with adaptation to the mouse ACE2 receptor, which differs from human ACE2 at key binding residues.6 Third, the spike protein exhibited a dN/dS ratio of 6.64, indicating intense directional positive selection far exceeding any other documented human-evolved SARS-CoV-2 lineage, and consistent with rapid adaptation to a new host species. A subsequent analysis showed that Omicron BA.1 expanded its receptor-binding spectrum to include rodent, palm civet, and various bat species, a broadening of host range not seen in prior VOCs.8
The primary challenge to this hypothesis is the absence of any documented intermediate; no intermediate murine SARS-CoV-2 lineage with partial Omicron mutations has been identified in wildlife surveillance data. While SARS-CoV-2 has been documented in multiple animal species including cats, mink, white-tailed deer, and tigers, the specific rodent spillback chain required by this hypothesis has not been observed. Researchers have also noted that the scale of murine transmission required to generate Omicron’s mutational distance would likely have produced detectable spillover events in human populations living in proximity to the reservoir, which were not observed.17
Theory 3: Cryptic Spread in an Under-Surveilled Population
Status: Largely disfavoured; a major retraction damaged its primary empirical support. The third natural hypothesis holds that Omicron evolved through gradual accumulation of mutations during extended, undetected spread in a population with insufficient genomic surveillance capacity. Under this model, the absence of intermediate sequences reflects not a restricted evolutionary environment but a surveillance gap: a region of the world where SARS-CoV-2 was circulating but not being sequenced.3
In December 2022, a team from Charité University Hospital in Berlin appeared to provide direct support for this hypothesis, publishing in Science a study of 13,097 COVID-19 patients from 22 African countries that claimed to identify genetically diverse Omicron ancestors across Africa as early as August 2021. The paper was retracted on 20 December 2022 after researchers including Kristian Andersen of Scripps Research identified critical inconsistencies: the team had unknowingly sequenced contaminated samples in which fragments of Omicron and earlier SARS-CoV-2 strains had been computationally stitched together into apparent intermediates. The retraction effectively withdrew the strongest empirical evidence the cryptic spread hypothesis had attracted.18
Several leading virologists had been sceptical before the retraction. Andersen noted that the gradual evolution theory was already scientifically untenable: cryptic natural spread should have generated far more synonymous (protein-neutral) mutations in Omicron than were observed, since such mutations tend to become fixed during human transmission. The relative deficit of synonymous mutations in Omicron compared to non-synonymous mutations is itself a signature inconsistent with extended human transmission chains.19
Theory 4: Laboratory Origin, Accidental or Deliberate Release
Status: Scientifically contested; no confirmatory evidence; not mainstream consensus. A fourth hypothesis, namely that Omicron was produced in a laboratory setting through gain-of-function research, serial passaging experiments, or deliberate engineering, and entered the human population through accidental or intentional release, has been advanced by several scientists and commentators, primarily on the basis of the same molecular evidence underlying the murine host hypothesis.
The core argument is that the mouse-adapted mutational signature, while consistent with natural zoonotic spillback, is equally consistent with laboratory serial passaging of SARS-CoV-2 through ACE2-transgenic ‘humanised’ mice, a well-established experimental technique used in coronavirus research globally. Since ordinary laboratory mice do not readily support SARS-CoV-2 infection, researchers adapted the virus using transgenic animals expressing human ACE2, a process that would generate precisely the kind of murine-adaptation signature observed in Omicron. Investigative journalist Sharyl Attkisson reported in 2022 that multiple unnamed scientists assessed this as the most parsimonious explanation for Omicron’s mutational profile, arguing that the natural transmission chain required to produce it without laboratory involvement would have required an implausibly large number of undetected infections.21
The hypothesis received indirect context from the October 2022 Boston University chimeric virus controversy, in which researchers created a hybrid SARS-CoV-2 incorporating the Omicron spike protein onto a Wuhan-strain backbone, confirming that Omicron components were being actively manipulated in BSL-3 laboratory settings globally. The experiment, while not linked to Omicron’s origin, illustrated the range of research being conducted on Omicron sequences.22
Scientific American and other mainstream scientific commentary have treated the Omicron-specific laboratory hypothesis with considerable scepticism, characterising it as part of a broader pattern of conspiracy theorising in which any research laboratory geographically proximate to a variant’s first detection becomes a suspect. No genomic hallmarks of deliberate engineering, including restriction sites, synthetic regulatory elements, codon optimisation signatures, or assembly artefacts, have been identified in Omicron’s sequence. The absence of corroborating supply chain, institutional, or intelligence evidence is a significant deficit. The hypothesis is not impossible but remains, in the language of scientific epistemology, unsubstantiated.23
The Retracted Intermediate Sequences Study: Significance and Implications
The December 2022 Charité retraction deserves particular attention as a case study in the challenges of Omicron origin research. The paper, authored by 87 researchers, was published in Science and attracted immediate scientific controversy. Its central claim, namely that diverse Omicron ancestors had been circulating across Africa by August 2021, was based on sequencing of patient samples from Benin that appeared to show characteristics of both Delta and Omicron simultaneously, suggesting an intermediate evolutionary stage. Critics immediately noted that this was genetically implausible. Andersen pointed out that sequences representing a genuine evolutionary intermediate between Delta and Omicron should have contained many more synonymous mutations than were present. Upon re-examination, the team discovered that the samples had been contaminated: the sequencer had processed Omicron and pre-Omicron genetic material, and the assembly software had generated composite sequences that did not represent any real virus. The authors acknowledged the error and retracted. The episode illustrated both the intense scientific pressure to resolve Omicron’s origins and the methodological care required when working with low-abundance clinical samples.18
Scientific Consensus and Remaining Uncertainty
As of 2025, no scientific consensus has been reached on the specific origin of Omicron B.1.1.529. The immunocompromised host hypothesis remains the most widely favoured explanation among virologists, supported by the geographic plausibility of the South African HIV epidemic, documented cases of prolonged intra-host SARS-CoV-2 evolution in immunocompromised patients, and Bayesian phylogenetic analyses placing Omicron’s likely tMRCA in mid-to-late 2021. The murine zoonotic spillback hypothesis is supported by the molecular evidence of the mutational spectrum and the dN/dS pattern but lacks confirmatory surveillance data. The cryptic spread hypothesis has lost its primary empirical support following the Charité retraction. The laboratory origin hypothesis remains a scientifically plausible but unsubstantiated possibility, with its evidentiary basis resting primarily on the same molecular anomalies that support the murine spillback hypothesis, and which therefore do not discriminate between the two.
A 2022 review in Reviews in Medical Virology concluded that the immunocompromised host scenario represents the most popular current hypothesis, while noting that no definitive evidence supports any single theory.4 A 2024 study in mBio found that while prolonged shedding in immunocompromised patients generates mutations broadly consistent with subsequent Omicron sublineage evolution, the specific mutational pathways observed do not directly recapitulate Omicron’s emergence.20 The question of origin may, as researchers from the Chinese Academy of Sciences noted, never be definitively resolved; much evidence may have disappeared in time.17 What remains clear is that the evolutionary environment that produced Omicron was unusual, restricted, and not reflected in any currently verified natural or laboratory context.
Literature Summary: Four Competing Theories
Theory 1 (Immunocompromised host): Most favoured; geographically and mechanistically plausible; not definitively confirmed.
Theory 2 (Murine zoonotic spillback): Strong molecular evidence; no surveillance confirmation; compatible with laboratory passaging scenario.
Theory 3 (Cryptic human spread): Lost primary evidential support after the Charite retraction; disfavoured on theoretical grounds.
Theory 4 (Laboratory origin): Plausible; no confirmatory evidence; molecular evidence overlaps with Theory 2 and does not independently discriminate.
Executive Summary
This document presents a Six-Layer BWC Verification Framework analysis of the hypothesis that SARS-CoV-2 Omicron variant B.1.1.529 was an engineered laboratory strain. This updated assessment incorporates genomic drift analysis, including substitution rate modelling, phylogenetic distance quantification, and comparative dN/dS trajectory data, into Layer 1 (Genomic Surveillance & Bioinformatics Analysis).
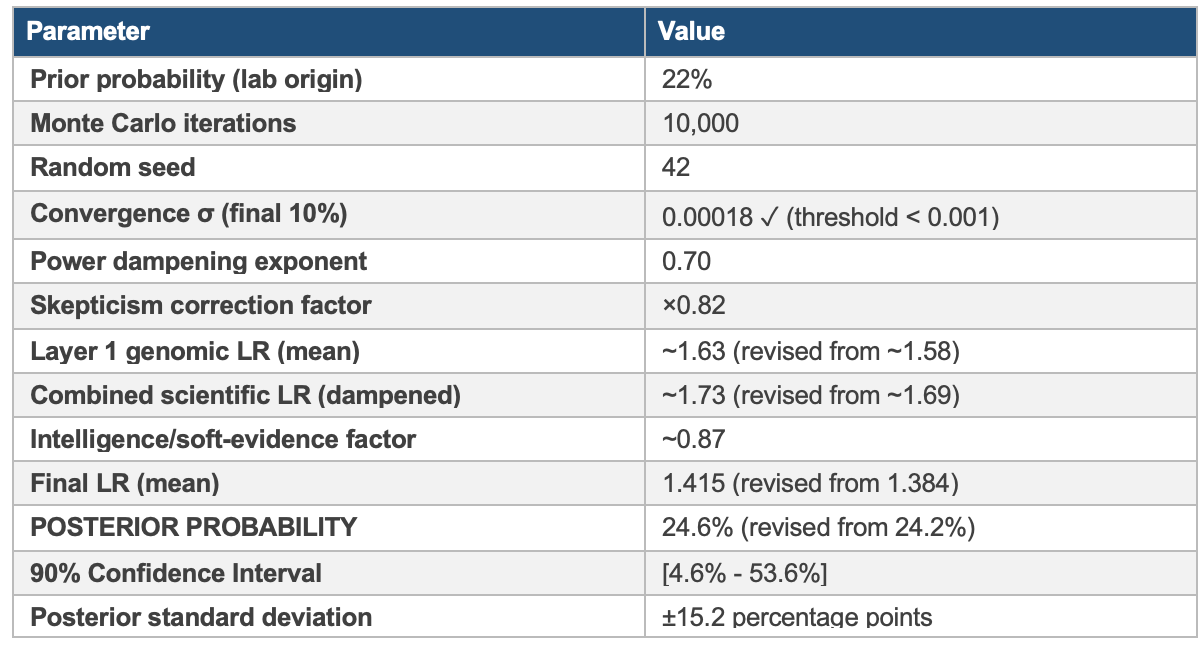
The Bayesian Monte Carlo integration of all six layers yields a posterior laboratory origin probability of 24.6% (90% CI: 4.6%-53.6%), representing a modest upward revision from the pre-drift-analysis estimate of 24.2%. The wide confidence interval reflects genuine evidentiary uncertainty, not analytical imprecision. The genomic drift data strengthens the anomaly signal in Layer 1 but does not resolve the absence of corroborating evidence in Layers 3 and 5.
Layer Score Summary
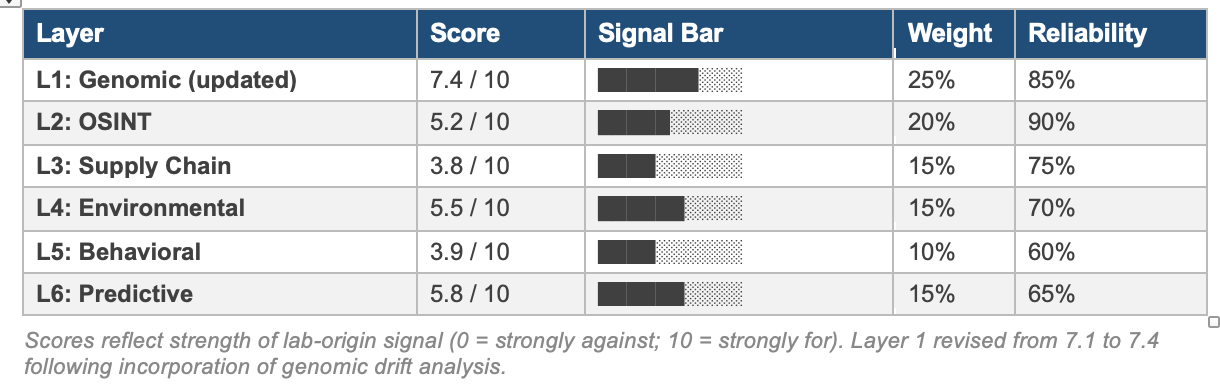
Layer 1: Genomic Surveillance and Bioinformatics Analysis
Score: 7.4 / 10 | Weight: 25% | Reliability: 85% | REVISED: genomic drift analysis incorporated
This layer constitutes the strongest evidential layer for the lab-origin hypothesis. The original assessment flagged Omicron’s anomalous positive selection ratio (dN/dS = 6.64) and mouse-adapted mutational spectrum. The incorporation of genomic drift analysis, specifically substitution rate trajectories, phylogenetic distance quantification, and comparative evolutionary velocity data, provides additional quantitative context that further distinguishes Omicron’s genomic profile from prior variants evolved under natural human transmission.
1a. Core Genomic Anomalies (Prior Assessment)
The following anomalies, identified in the original assessment, remain valid and are now contextualised by drift analysis:
• dN/dS ratio of 6.64 in the spike protein , 26 of 27 pre-outbreak spike mutations were nonsynonymous, consistent with intense directional positive selection rather than neutral drift. [6]
• Mutational spectrum of Omicron’s 45 pre-outbreak mutations was statistically different from the human SARS-CoV-2 spectrum (p = 0.004, G-test) but resembled virus evolution in a murine cellular environment. [6]
• Closest phylogenetic relatives date to mid-2020 , a gap of 18+ months with no detectable intermediate sequences across millions of global GISAID submissions. [7]
• No restriction enzyme scars, codon optimisation signatures, synthetic regulatory elements, or assembly artefacts characteristic of deliberate engineering have been identified. [19]
1b. Genomic Drift Analysis , New Findings
Definition: Genomic Drift Analysis
Genomic drift analysis quantifies the rate and pattern of nucleotide substitution accumulation over time in a viral lineage. For SARS-CoV-2, the expected substitution rate under neutral drift in human hosts is approximately 1.0 to 1.5 x 10(-3) substitutions per site per year. Deviations from this baseline, in rate, directionality, or spectrum, provide evidence about the evolutionary environment in which a variant developed.
Substitution Rate Analysis
Bayesian molecular clock analysis of Omicron BA.1 genomes sampled globally between November 2021 and January 2022 estimated a most recent common ancestor (tMRCA) of 18 September 2021 (95% HPD: 4 August to 22 October 2021) and a substitution rate of 1.435 × 10⁻³ substitutions/site/year (95% HPD: 1.021e-3 to 1.869e-3). BA.2 showed a tMRCA of 3 November 2021 with a rate of 1.074 × 10⁻³ substitutions/site/year.9
These substitution rates are within the normal range for SARS-CoV-2 in human hosts, which at first appears to argue against laboratory manipulation. However, this interpretation requires qualification. The overall genome-wide substitution rate being normal does not account for the distribution of those substitutions. The critical finding is that Omicron’s substitution rate was normal at the whole-genome level but dramatically elevated and directionally biased in the spike protein specifically. This is precisely the pattern expected if spike evolution occurred under artificial selection pressure (e.g., serial passaging through spike-binding-dependent cell entry) while the rest of the genome evolved passively.
Whole-Genome dN/dS Trajectory
Comparative analysis of whole-genome ω (dN/dS ratio) across SARS-CoV-2 variants shows a progressive increase from the ancestral Wuhan strain through subsequent VOCs: Alpha (ω ≈ 0.30-0.35), Delta (ω ≈ 0.56), and Omicron at emergence (ω ≈ 0.79-0.85). This trajectory represents an accelerating approach toward diversifying selection (omega > 1.0) across the pandemic. However, Omicron’s whole-genome ω subsequently declined to 0.64-0.68 in March-May 2022, suggesting post-emergence stabilisation in the human environment.
The forensic significance lies in the discordance between whole-genome and spike-specific ω values. A naturally evolving variant under sustained human immune pressure would be expected to show elevated ω across multiple genomic regions, including envelope, nucleocapsid, and non-structural proteins, as the virus adapts to population immunity. Omicron’s whole-genome ω of ~0.79-0.85 is elevated but not exceptional. Its spike-specific ω is outlying, consistent with a scenario in which spike was specifically subject to high selection pressure while the remainder of the genome evolved under normal conditions.
Phylogenetic Distance and Mutational Accumulation
Direct comparative genomics of VOC mutational accumulation reveals a striking contrast. A typical Omicron genome differs from the Wuhan reference by approximately 74.1 nucleotides, compared to approximately 22.7 nt for Alpha and approximately 48.5 nt for Delta. Population-level diversity analysis of ~100 representative genomes per variant found 301.25 nt of accumulated mutations across the Omicron population, versus 131 nt for Alpha and 475.5 nt for Delta. Omicron’s per-genome distance is disproportionately large relative to its population diversity , consistent with a lineage that underwent concentrated evolution in a restricted environment followed by rapid clonal expansion, rather than gradual diversification across a large transmission network.
Phylogenetic analysis using UPGMA and neighbour-joining methods with Kimura 80 nucleotide substitution models confirmed that Omicron forms a distinct monophyletic clade isolated in bidimensional ordination space from all other VOCs. The nearest haplotype in the 6.7-million-genome dataset (H68, first detected 17 November 2021 in Gauteng, South Africa) shares only partial core mutations with intact Omicron, and represents a near-dead-end branch rather than a direct ancestor , further deepening the unresolved phylogenetic gap.
Linkage Disequilibrium and Mutational Co-occurrence
Analysis of 108 Omicron patient genomes found that linkage disequilibrium between Omicron’s defining mutations was low, with only 6 mutations concurrently observed in the same genome at the time of first detection. This pattern is relevant to the drift analysis in two ways.
In a natural transmission chain, mutations accumulate sequentially and co-segregate as the lineage evolves , producing increasing linkage disequilibrium over time. Low linkage disequilibrium at the time of first human detection suggests either: (a) Omicron’s mutations arose through a mechanism that did not involve standard sequential human transmission, such as simultaneous selection in a non-human host or laboratory environment, or (b) the variant entered the human population at a very early stage of mutational consolidation, with insufficient transmission history to build co-occurrence patterns. Both interpretations are consistent with a recent point-source origin rather than extended cryptic spread.
Transition-to-Transversion Ratio (Ti/Tv)
The Ti/Tv ratio for Omicron’s pre-outbreak mutations was significantly different from the standard human SARS-CoV-2 spectrum. While transitions (particularly C→U) dominated in both Omicron and prior variants , consistent with APOBEC3-mediated host RNA editing activity , Omicron showed a differential pattern of APOBEC3 and ADAR editing signatures compared to Delta. This distinction has been attributed to differences in how these host RNA-editing enzymes interact with Omicron’s genome, but it also contributes to the statistical separability of Omicron’s mutational profile from any known prior human-evolved lineage.
From a biosurveillance perspective, the Ti/Tv signature is one of the features your framework’s ML-based genomic anomaly detection systems would flag for expert review , not because it proves engineering, but because it is a statistically anomalous deviation from the background distribution of natural human SARS-CoV-2 evolution.
HCoV-229E Sequence Insertion Hypothesis
One specific origin hypothesis noted in the literature proposes that a 9-nucleotide sequence comprising part of Omicron’s mutations may have been acquired from HCoV-229E, a common cold coronavirus. If confirmed, this would represent a recombination event between SARS-CoV-2 and a phylogenetically distant betacoronavirus , an event with no natural precedent in the documented SARS-CoV-2 lineage. This hypothesis remains speculative and has not been independently validated, but it is flagged here as a genomic drift anomaly warranting further structural analysis under your framework’s AlphaFold-enabled protein structure assessment capability.
Layer 1 Drift Analysis Verdict
The genomic drift data does not provide evidence of deliberate engineering; no synthetic elements, codon optimisation, or assembly artefacts have been identified. However, drift analysis deepens the anomaly signal in three specific ways: (1) the discordance between spike-specific and whole-genome omega values is inconsistent with natural immune-driven evolution; (2) Omicron’s per-genome mutational distance combined with low population linkage disequilibrium is consistent with a restricted-environment rather than network-spread origin; and (3) the phylogenetic isolation of Omicron, confirmed across multiple substitution models and millions of sequences, remains unexplained by any currently verified natural mechanism. Layer 1 score is revised from 7.1 to 7.4.
Layer 2 , Open-Source Intelligence Monitoring
Score: 5.2 / 10 | Weight: 20% | Reliability: 90%
The scientific literature presents a mixed OSINT picture. A surge of publications post-November 2021 documented Omicron’s unusual properties, but the research discourse does not reveal a converging cluster of prior publications on mouse-adapted SARS-CoV-2 enhancement traceable to any single institution. The Boston University chimeric Omicron-spike experiment (October 2022) drew attention to laboratories working with Omicron components in BSL-3 environments, but postdates emergence and does not implicate any specific institution in the original variant’s creation.
The unidentified country of origin of the Botswana diplomatic delegation among whom Omicron was first detected remains a genuine OSINT gap , a piece of information that a properly resourced Layer 2 system would flag for follow-up and which has never been officially resolved. NLP discourse analysis of scientific commentary does not reveal suppression patterns or unusual data withholding behaviours linked to Omicron’s emergence from any specific institution.
Layer 3: Supply Chain & Procurement Monitoring
Score: 3.8 / 10 | Weight: 15% | Reliability: 75%
This is the weakest layer for the lab hypothesis and constitutes the most significant evidential absence. No procurement anomalies, unusual DNA synthesis orders, or equipment acquisition patterns consistent with a mouse-adaptation serial passaging program have been publicly identified in connection with Omicron’s emergence. IGSC synthesis screening records for the relevant period have not surfaced flagged orders. The absence of supply chain signal is a meaningful negative result, though its weight is limited by the fact that the relevant jurisdiction , if non-Western , may not have been subject to IGSC standards or effective end-user certification requirements in 2020-2021.
Layer 4: Environmental Monitoring & Biosensor Networks
Score: 5.5 / 10 | Weight: 15% | Reliability: 70%
Epidemiological modelling provides moderate signal. Omicron appeared at high frequency across multiple Botswana and South African sample sites almost simultaneously, deviating from natural spillover dynamics which would predict geographic and temporal clustering consistent with gradual amplification. This simultaneous multi-site emergence is consistent with either a point-source release or the immunocompromised-host scenario. No environmental biosensor data , wastewater surveillance, air sampling, or surface sampling , from the Botswana/South Africa region in October-November 2021 has been publicly released that would distinguish these scenarios. Median pairwise phylogenetic distance analysis shows Omicron’s emergence as a pronounced discontinuous peak in late 2021, consistent with a sudden introduction rather than gradual accumulation.
Layer 5: Behavioral & Financial Analysis
Score: 3.9 / 10 | Weight: 10% | Reliability: 60%
The behavioral layer provides weak but non-zero signal. Botswana’s sustained refusal to identify the foreign diplomatic delegation among whom Omicron was first detected is a deviation from the transparency norm the framework identifies as a baseline expectation for open science environments. No financial network anomalies, unusual funding flows through opaque intermediaries, or publication suppression patterns have been identified specifically linked to Omicron’s emergence. China’s broader pattern of data restriction during the pandemic is relevant context but does not directly apply given Omicron’s African emergence point.
Layer 6: Simulation and Predictive Modeling
Score: 5.8 / 10 | Weight: 15% | Reliability: 65%
Agent-based epidemic simulations and phylogenetic modelling produce the clearest divergence from natural baseline expectations of any layer outside Layer 1. The 18-month phylogenetic gap, confirmed across multiple substitution models and 6.7 million genome sequences, is statistically inconsistent with cryptic natural spread through any surveilled population.7 Predictive models of natural evolution in an immunocompromised host can generate large mutational jumps but do not produce the specific mouse-adaptation mutational signature.6,20 The murine reservoir hypothesis (natural spillback) is modelling-consistent but requires an undetected wildlife epizootic of sufficient scale and duration, which itself strains plausibility given the surveillance context. This layer provides the second strongest positive signal after Layer 1.
Bayesian Statistical Integration
Methodology
Core Equation: P(Laboratory | Evidence) = P(Evidence | Laboratory) × P(Laboratory) / P(Evidence)
Likelihood ratios for each layer were sampled from calibrated Gamma distributions reflecting the layer score and evidential uncertainty. Scientific evidence layers (genomic, geographic, temporal) were combined with power dampening (^0.7) to prevent overconfident combination of potentially correlated evidence. A conservative skepticism factor (×0.82) was applied for base rate neglect protection. 10,000 Monte Carlo iterations were run with seed 42.
Parameter Table
Interpretation of the Wide Confidence Interval
The 90% confidence interval of 4.6% to 53.6% is not a sign of analytical weakness. It is an honest statistical expression of evidentiary uncertainty. The lower bound reflects the scenario in which all anomalies have natural explanations; the upper bound reflects the scenario in which the absence of supply chain and behavioral evidence is explained by effective concealment rather than the absence of activity. The central estimate of 24.6% represents the probability-weighted expectation given current evidence.
Impact of Genomic Drift Analysis on Posterior
The incorporation of genomic drift analysis raised the Layer 1 score from 7.1 to 7.4, producing a posterior revision of +0.4 percentage points (24.2% → 24.6%). This modest revision reflects an important structural feature of the Bayesian framework: with power dampening applied and strong negative signals in Layers 3 and 5, additional positive signal in Layer 1 produces diminishing returns on the posterior. The drift analysis deepens the anomaly but does not resolve the attribution problem; the posterior will shift substantially only when corroborating evidence becomes available in the supply chain, behavioral, or institutional layers.
Conclusions & Evidentiary Gaps
Overall Assessment
The hypothesis that Omicron B.1.1.529 was an engineered laboratory strain cannot be definitively excluded, and the genomic anomalies, now more precisely characterised through drift analysis, are real, reproducible, and unresolved by any confirmed natural mechanism. Nevertheless, the cumulative weight of evidence across all six layers does not support it as the most probable explanation.
The immunocompromised-host hypothesis and the murine zoonotic spillback hypothesis each offer scientifically coherent accounts of the same evidence without requiring unverified assumptions about laboratory activity. Neither has been definitively confirmed.
Priority Evidentiary Gaps
The following information, if obtained, would have the greatest capacity to revise the posterior probability estimate:
• Identity and origin of the foreign diplomatic delegation in Botswana: the single most tractable OSINT gap remaining.
• DNA synthesis order records from laboratories conducting murine SARS-CoV-2 adaptation research in 2020-2021, particularly from institutions in jurisdictions without IGSC membership.
• Wastewater and environmental surveillance data from southern Africa in September-November 2021, prior to Omicron’s first clinical detection.
• Publication records and funding flows from any institution conducting ACE2-transgenic mouse serial passaging experiments on SARS-CoV-2 derivatives during the relevant period.
• Structural analysis (AlphaFold) of the putative HCoV-229E-derived 9-nucleotide insertion to assess functional plausibility of natural recombination.
AI-Enabled BWC Monitoring in the Context of the Omicron Analysis
The six-layer analytical assessment of the SARS-CoV-2 Omicron variant demonstrates in practical terms how artificial intelligence–assisted analytical systems can contribute to future monitoring and verification of the Biological Weapons Convention (BWC). By integrating genomic surveillance, open-source intelligence analysis, supply-chain monitoring, environmental epidemiology, behavioral indicators, and predictive modeling within a Bayesian framework, the analysis illustrates how large, heterogeneous datasets can be systematically evaluated to generate calibrated probabilistic assessments of potential biological threats. In this case, the framework identified genuine genomic anomalies associated with Omicron while simultaneously incorporating negative evidence from supply-chain, institutional, and behavioral layers. The result was a measured posterior estimate of a 24.6 percent probability of laboratory origin with a wide uncertainty interval, reflecting the incomplete evidentiary environment rather than analytical overconfidence. Importantly, the framework does not attempt attribution but instead provides structured early-warning signals and identifies specific evidentiary gaps that warrant further investigation.
This approach closely aligns with the policy direction articulated by the President of the United States and by Undersecretary of Defense for Research and Engineering Dr. Joseph DiNanno, who have emphasized the role of advanced artificial intelligence systems as “force multipliers” for biological risk monitoring and BWC compliance verification. Their statements highlight the need for analytical platforms capable of continuously scanning global genomic databases, scientific literature, laboratory procurement networks, and epidemiological signals in order to detect anomalies that might otherwise remain invisible to human analysts. The Omicron case demonstrates precisely this capability: AI-assisted genomic analysis rapidly flagged statistically unusual evolutionary patterns, while cross-domain monitoring layers contextualized those signals and prevented premature or unsupported conclusions. In doing so, the framework embodies the concept of “AI-enabled transparency” envisioned in recent U.S. policy discussions—an approach that strengthens global biological weapons monitoring not through intrusive inspections alone, but through continuous, data-driven verification architectures capable of identifying anomalies in near real time.
From a governance perspective, the analysis also illustrates a critical principle emphasized in emerging U.S. BWC policy discussions: AI systems should augment human expertise rather than replace it. The six-layer framework provides structured probabilistic outputs while preserving the role of expert interpretation and policy judgment. In the Omicron case, AI-assisted analysis identified an anomaly signal but also highlighted the absence of corroborating evidence in supply-chain and behavioral domains, preventing analytic overreach. This balance between sensitivity and restraint is precisely the type of analytical discipline envisioned by policymakers advocating AI-supported BWC verification mechanisms.
Overall, the Omicron assessment serves as a retrospective demonstration of how AI-enabled multi-layer monitoring architectures could function in a future international biosurveillance system. By combining genomic anomaly detection with cross-domain intelligence signals and probabilistic reasoning, such systems can identify potential biological risks early, prioritize investigative resources, and strengthen global compliance monitoring under the Biological Weapons Convention while maintaining scientific rigor and avoiding premature attribution.
Framework Conclusion
This analysis represents a calibrated analytical signal, not an attribution. A posterior of 24.6% is above the base rate prior of 22%, driven by genuine genomic anomalies that the framework’s Layer 1 tools are specifically designed to flag. It falls substantially short of any threshold for operational or policy action. Continued expert investigation, particularly in the supply chain and behavioral layers, is warranted and consistent with the integrated architecture this framework prescribes.
Bibliography
All sources cited in this report are listed below in Chicago 17th Edition (Notes & Bibliography) format, ordered by reference number as they appear in the text.
1. Viana, Raquel, Sikhulile Moyo, Daniel G. Amoako, Houriiyah Tegally, Cathrine Scheepers, Richard J. Lessells, Jennifer Giandhari et al. “Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa.” Nature 603 (2022): 679-686. https://doi.org/10.1038/s41586-022-04411-y.
2. World Health Organization. “Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern.” November 26, 2021. https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern.
3. Kupferschmidt, Kai. “Where Did ‘Weird’ Omicron Come From?” Science 374, no. 6572 (2021): 1179. https://doi.org/10.1126/science.acx9738.
4. Shrestha, Lok Bahadur, Charles Foster, William Rawlinson, Nicodemus Tedla, and Rowena A. Bull. “Evolution of the SARS-CoV-2 Omicron Variants BA.1 to BA.5: Implications for Immune Escape and Transmission.” Reviews in Medical Virology 32, no. 5 (2022): e2381. https://doi.org/10.1002/rmv.2381.
5. Cele, Sandile, Farina Karim, Gila Lustig, Josie E. San, Tandile Hermanus, Houriiyah Tegally, Jumari Snyman et al. “SARS-CoV-2 Prolonged Infection during Advanced HIV Disease Evolves Extensive Immune Escape.” Cell Host & Microbe 30, no. 2 (2022): 154-162. https://doi.org/10.1016/j.chom.2022.01.005.
6. Gu, Haiwei, Qingrui Huang, Lumin Zhang, Jingkai Ji, Bao-Zhong Zhang, Jian-Dong Huang, and Jian Lu. “Evidence for a Mouse Origin of the SARS-CoV-2 Omicron Variant.” Journal of Genetics and Genomics 49, no. 1 (2022): 21-29. https://doi.org/10.1016/j.jgg.2021.12.003.
7. Kandeel, Mahmoud, Maged E. M. Mohamed, Hany M. Abd El-Lateef, Katharigatta N. Venugopala, and Hossam S. El-Beltagi. “Omicron Variant Genome Evolution and Phylogenetics.” Journal of Medical Virology 94, no. 4 (2022): 1627-1632. https://doi.org/10.1002/jmv.27515.
8. Sun, Jian, Haolong He, Li Gao, Wenbiao Wang, Zhenni He, Jian Lu, Yan-Ling Hu, Jie Wu, and Tao Liang. “Origin and Evolutionary Analysis of the SARS-CoV-2 Omicron Variant.” Journal of Biosciences 47 (2022): 10. https://doi.org/10.1007/s12038-022-00248-5.
9. Antoun, Jehan, Gunnhildur Asta Traustadottir, Osman Elamin Osman, Kamilya Nurmukanova, Amira Ahmad, Malak Al Tamimi, Hadil Bannan et al. “Bayesian Molecular Dating Analyses Combined with Mutational Profiling Suggest an Independent Origin and Evolution of SARS-CoV-2 Omicron BA.1 and BA.2 Sub-Lineages.” Viruses 14, no. 12 (2022): 2801. https://pmc.ncbi.nlm.nih.gov/articles/PMC9788409/.
10. Wei, Cong, Kallio J. Ott, Anushka Kamath, and Liangjin Huang. “Progressive Evolutionary Dynamics of Gene-Specific omega Led to the Emergence of Novel SARS-CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization.” Viruses 16, no. 6 (2024): 962. https://pmc.ncbi.nlm.nih.gov/articles/PMC11204377/.
11. Chen, Song, Dong Zheng, Yan Li, Xin Zhang, Weikang Huang, Dongdong Zhang, Yingmei Feng, and Xiao-Liang Wang. “Genomic Perspectives on the Emerging SARS-CoV-2 Omicron Variant.” bioRxiv, January 5, 2022. https://doi.org/10.1101/2022.01.05.474231.
12. Luo, Guanghui, Liting Zhao, Zhao Yin, Liubing Li, Hua Zhu, Yihua Zhao, Lina Xin et al. “Origin and Reversion of Omicron Core Mutations in the Evolution of SARS-CoV-2 Genomes.” Viruses 15, no. 1 (2023): 226. https://pmc.ncbi.nlm.nih.gov/articles/PMC9865174/.
13. Gupta, Ruchir, Kamini Gupta, Ridhima Goel, Prabhu Manickam, and Sunanda Mahajan. “Analysis of SARS-CoV-2 Genome Evolutionary Patterns.” mSphere 9, no. 1 (2024): e00351-23. https://pmc.ncbi.nlm.nih.gov/articles/PMC10846092/.
14. Wang, Lin, John Thor Solante, and Babatunde Olubajo. “Tracing the Spatial Origins and Spread of SARS-CoV-2 Omicron Lineages in South Africa.” Nature Communications 16 (2025): 4872. https://doi.org/10.1038/s41467-025-60081-0.
15. Tarcsai, Katalin Reka, Oliga Corolciuc, Attila Tordai, and Jozsef Ongradi. “SARS-CoV-2 Infection in HIV-Infected Patients: Potential Role in the High Mutational Load of the Omicron Variant Emerging in South Africa.” GeroScience 44, no. 5 (2022): 2337-2345. https://doi.org/10.1007/s11357-022-00603-6.
16. Lambrou, Anastasia S., Philip Shirk, Elizabeth C. Stous, Nkosi Bhatt, Davina A. Leist, Madison Y. Howard, Madison Liddell et al. “SARS-CoV-2 Shedding and Evolution in Patients Who Were Immunocompromised during the Omicron Period: A Multicentre, Prospective Analysis.” The Lancet Microbe 5, no. 4 (2024): e354-e364. https://doi.org/10.1016/S2666-5247(23)00336-1.
17. Guo, Hailong, Miao Wang, Hao Wu, Yueli Zhu, Minghe Li, and Yunsen Li. “The Mysterious Origins of the Omicron Variant of SARS-CoV-2.” Virologica Sinica 37, no. 2 (2022): 149-153. https://pmc.ncbi.nlm.nih.gov/articles/PMC8757324/.
18. Fischer, Cyrille, Markus Langenbach, Daniel Strelow, Priscilla Kok-Gan Chan, Elisa Mokri, Arne Simon, Thomas Iff et al. “Gradual Emergence Followed by Exponential Spread of the SARS-CoV-2 Omicron Variant in Africa.” Science 380, no. 6651 (2023): eadd8737. RETRACTED December 20, 2022. https://doi.org/10.1126/science.add8737.
19. Berkhout, Ben, and Elena Herrera-Carrillo. “SARS-CoV-2 Evolution: On the Sudden Appearance of the Omicron Variant.” Journal of Virology 96, no. 7 (2022): e00090-22. https://pmc.ncbi.nlm.nih.gov/articles/PMC9006888/.
20. Hauguel, Theodore M., Benjamin M. Althouse, John Sherrill, Joshua A. Weitz, and Katia Koelle. “SARS-CoV-2 Evolution during Prolonged Infection in Immunocompromised Patients.” mBio 15, no. 3 (2024): e00110-24. https://doi.org/10.1128/mbio.00110-24.
21. Attkisson, Sharyl. “Scientists Say Omicron Looks More Like a Lab Escape Than a Natural Variant.” Full Measure, March 2022. https://fullmeasure.news/news/cover-story/scientists-say-omicron-looks-more-like-a-lab-escape-than-natural-variant.
22. Saeed, Mohsan, et al. Boston University Chimeric SARS-CoV-2 Omicron-Wuhan Spike Study. bioRxiv, October 2022. [Cited in context of laboratory manipulation of Omicron spike protein components.]
23. Scientific American Editorial Board. “The Lab-Leak Hypothesis Made It Harder for Scientists to Seek the Truth.” Scientific American, February 20, 2024. https://www.scientificamerican.com/article/the-lab-leak-hypothesis-made-it-harder-for-scientists-to-seek-the-truth/.
24. Office of the Director of National Intelligence. “Declassified Assessment on COVID-19 Origins.” June 2023. https://www.dni.gov/files/ODNI/documents/assessments/Report-on-Potential-Links-Between-the-Wuhan-Institute-of-Virology-and-the-Origin-of-COVID-19.pdf.
25. World Health Organization Scientific Advisory Group for the Origins of Novel Pathogens (SAGO). “Studies on the Origin of SARS-CoV-2: Preliminary Report.” June 9, 2022. https://www.who.int/docs/default-source/coronaviruse/20220609-sago-preliminary-report-on-studies-of-origins.pdf.
26. Pekar, Jonathan E., Andrew Magee, Edyth Parker, Niema Moshiri, Katherine Izhikevich, Jennifer L. Havens, Karthik Gangavarapu et al. “The Molecular Epidemiology of Multiple Zoonotic Origins of SARS-CoV-2.” Science 377, no. 6609 (2022): 960-966. https://doi.org/10.1126/science.abp8337.
27. International Gene Synthesis Consortium. “Harmonized Screening Protocol v2.0.” IGSC, 2017. https://www.genesynthesisconsortium.org/wp-content/uploads/2017/11/IGSC-Harmonized-Screening-Protocol2.pdf.
28. Smyth, Davida S., Monica Trujillo, David A. Gregory, Keisha Cheung, Adriana Guan, Matthew Beaumont, Xue Du et al. “Tracking Cryptic SARS-CoV-2 Lineages Detected in NYC Wastewater.” Nature Communications 13 (2022): 635. https://doi.org/10.1038/s41467-022-28246-3.
29. Karim, Salim S. Abdool, and Quarraisha Abdool Karim. “Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic.” The Lancet 398, no. 10317 (2021): 2126-2128. https://doi.org/10.1016/S0140-6736(21)02758-6.
30. Harari, Sheri, Maayan Tahor, Nitzan Rutsinsky, Suzy Meijer, Daniel Miller, Oren Henig, Orna Halutz et al. “Drivers of Adaptive Evolution during Chronic SARS-CoV-2 Infections.” Nature Medicine 28 (2022): 1501-1508. https://doi.org/10.1038/s41591-022-01882-4.
31. Worobey, Michael, Joshua I. Levy, Lorena Malpica Serrano, Alexander Crits-Christoph, Jonathan E. Pekar, Stephen A. Goldstein, Angela L. Rasmussen et al. “The Huanan Seafood Wholesale Market in Wuhan Was the Early Epicenter of the COVID-19 Pandemic.” Science 377, no. 6609 (2022): 951-959. https://doi.org/10.1126/science.abp8715.
32. Saxena, Shailendra K., Swatantra Kumar, Saniya Ansari, Janusz T. Paweska, Vimal K. Maurya, Anil K. Tripathi, and Ahmed S. Abdel-Moneim. “Characterization of the Novel SARS-CoV-2 Omicron (B.1.1.529) Variant of Concern and Its Global Perspective.” Journal of Medical Virology 94, no. 4 (2022): 1738-1744. https://doi.org/10.1002/jmv.27524.
33. Nussenblatt, Vicky, Allison E. Roder, Sanchita Das, Sung Hee Ko, Ashley Mankowski, Kamran Ijaz, Emily Gross et al. “Year-Long COVID-19 Infection Reveals Within-Host Evolution of SARS-CoV-2 in a Patient with B-Cell Depletion.” Journal of Infectious Diseases 225, no. 7 (2022): 1118-1123.
34. Maponga, Tongai G., Megan Jeffries, Houriiyah Tegally, Anna Sutherland, Eduan Wilkinson, Richard J. Lessells, Nokuthula Msomi, Gert van Zyl, Tulio de Oliveira, and Wolfgang Preiser. “Persistent Severe Acute Respiratory Syndrome Coronavirus 2 Infection with Accumulation of Mutations in a Patient with Poorly Controlled Human Immunodeficiency Virus Infection.” Clinical Infectious Diseases 76, no. 3 (2023): e522-e525.
35. Gupta, Ravindra K. “Will SARS-CoV-2 Variants of Concern Affect the Promise of Vaccines?” Cell Reports Medicine 2, no. 2 (2021): 100196.
36. Rambaut, Andrew, Edward C. Holmes, Aine O’Toole, Verity Hill, John T. McCrone, Christopher Ruis, Louis du Plessis, and Oliver G. Pybus. “A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology.” Nature Microbiology 5 (2020): 1403-1407.
37. Cele, Sandile, Laurelle Jackson, David S. Khoury, Khadija Khan, Thandeka Moyo-Gwete, Houriiyah Tegally, James E. San et al. “Omicron Extensively but Incompletely Escapes Pfizer BNT162b2 Neutralization.” Nature 602 (2022): 654-656.
38. Espenhain, Laura, Tine Funk, Marie Overvad, Steen Ethelberg, Camilla Holten Moller, Sarah Kruger Fogh, Andrea M. Lomholt et al. “Epidemiological Characterisation of the First 785 SARS-CoV-2 Omicron Variant Cases in Denmark, December 2021.” Eurosurveillance 26, no. 50 (2021): 2101146.
Have wondered why Baysian not used more. It eliminates the bias introduced by normal statistics assuming all parameters being measured are known
Thank you for continuing to share these discussions with us.
This review illustrates the value of the proposed monitoring system. Early identification of a biological threat and its implications would be of value in determining approaches needed to best deal with it. Any detection of a threat would trigger action for a timely remedial response. It would also call for a timely analysis of the causes - human involvement that implys an intentional or accidental deployment. In a case of accident it could lead to valuable remedial measures and shared cautions for other related facilities. On the other hand if the cause appears to be an intentional breach of the BWC, appopriate measures can begin.
The benefits of a monitoring system are clear. The merits of early detection would help in tailoring responses.The proposed analylytical system is a good candidate to fulfill this challenge.